Genetics & Your Health
Genetics plays a powerful role in shaping your health. For certain conditions, genetic testing and counseling can help you better understand the role that genetics plays and empower you with the information you need to manage your health.
Genetics & Metabolic Disorders
Our metabolism is a complex process in which our bodies produce energy from the food and drink that we take in. This process occurs through a series of steps. Metabolism also involves the removal of waste that is produced during this process. Metabolic disorders refer to a group of genetic conditions in which there is an error somewhere in this process. Specifically, an individual is not able to produce or break down certain enzymes, proteins, and/or other substances needed by the body. For this reason, metabolic disorders are also called biochemical disorders or inborn errors of metabolism.
There are hundreds of different kinds of metabolic disorders. The disorders are categorized by the specific process or pathway that they affect. The primary categories include:
- Amino acid disorders
- Carbohydrate disorders
- Fatty acid oxidation disorders
- Lysosomal disorders (e.g Fabry disease)
- Mitochondrial disorders
- Organic acid disorders
- Peroxisomal disorders
- Urea cycle disorders (e.g. OTC deficiency)
- Congenital disorders of creatine metabolism
- Congenital disorders of glycosylation
- Lipid metabolism disorders (e.g. familial hypercholesterolemia)
- Metal metabolism disorders (e.g. Wilson disease)
- Purine and pyrimidine metabolism disorders
- Porphyrin metabolism disorder
- Vitamin and cofactor metabolism disorder
Metabolic disorders may affect a single part of the body, or multiple body systems, depending on what specific pathway is involved. Individuals may have limited to no symptoms or may present with life-threatening episodes. In addition, symptoms may vary between infancy to childhood to adulthood, can be progressive, and at times life-limiting. A metabolic disorder may be suspected in people with developmental disabilities, seizures, poor growth, and periods of regression.
Diagnosis of a metabolic disorder is made through the careful evaluation of an individual’s medical and family history, physical examination, and measurement of specific levels of certain substances in the blood and urine. In some cases, imaging studies, measurement of levels of certain substances in other body tissues (e.g. skin or muscle), and/or genetic testing may be recommended.
For many metabolic disorders, there is specific management and treatment available. In general, treatment is focused on reducing and eliminating the buildup of these toxic substances in the body. This may be done through a specialized diet, taking supplements, or avoidance of triggers, and some metabolic conditions can now even be treated by replacing the missing enzymes.
Genetics & Your Brain
Controlling more than just your thoughts, your brain is involved in functions that seem simple, such as eating and breathing, as well as functions that are complex like your interactions with the wider world, processing emotions, and learning new information. While many people think about genes contributing to our physical features, such as eye color or height, some genes influence how our brain develops and continues to function during our lifetime. These genes can influence how your brain is shaped, how you develop, how you process and interact with the world around you, and how your brain communicates with your body. These genes help control the internal mechanics that build all of the tissues in our brain and then how the brain communicates through neurons and the internal network.
Genetic conditions that affect the brain may do so in a variety of ways. These conditions may impact the overall development, structure, or size of the brain to whether the brain is able to produce or utilize important proteins to function and communicate to the many interactions with the other factors to form complex conditions like dementia and Autism spectrum disorder to much more that we are still uncovering.
As researchers continue to delve into the mysteries of genetics and your brain, here are different categories and examples of some genetic brain conditions:
- Neurodevelopmental
- Epilepsy
- Neurometabolic (e.g. phenylketonuria, PKU)
- Structural brain disorders (e.g. agenesis of the corpus callosum, holoprosencephaly, lissencephaly, microcephaly, schizencephaly)
- Neurodegenerative
- Huntington’s disease (HD)
- Amyotrophic Lateral Sclerosis (ALS)
- Frontotemporal dementia (FTD)
- Alzheimer’s disease
- Parkinson’s disease
- Leukodystrophies
- Psychiatric
Genetics & Your Heart
The heart is one of the more complex and vital organs in our body, responsible for pumping blood that carries oxygen all throughout our bodies via the blood vessels. To better understand health conditions that can affect our heart, it is important to first have an understanding of how the heart is supposed to work.
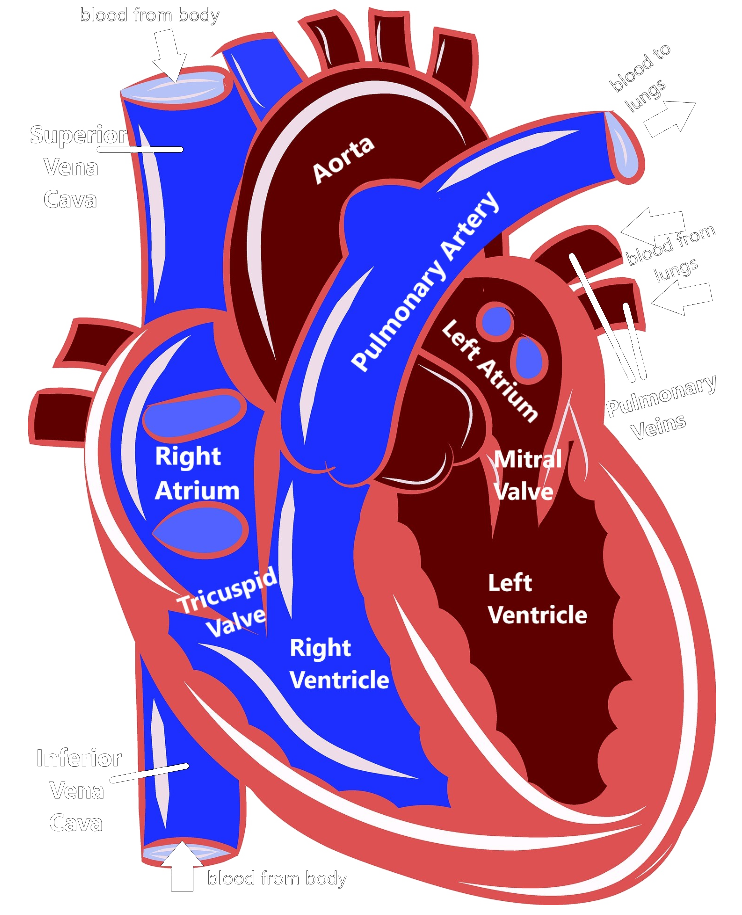
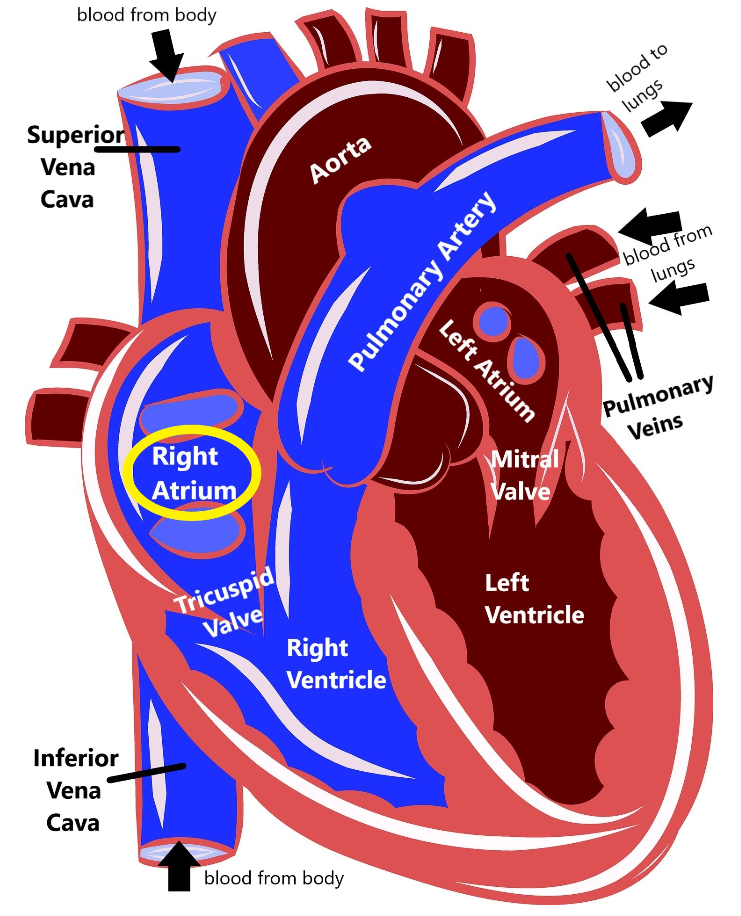
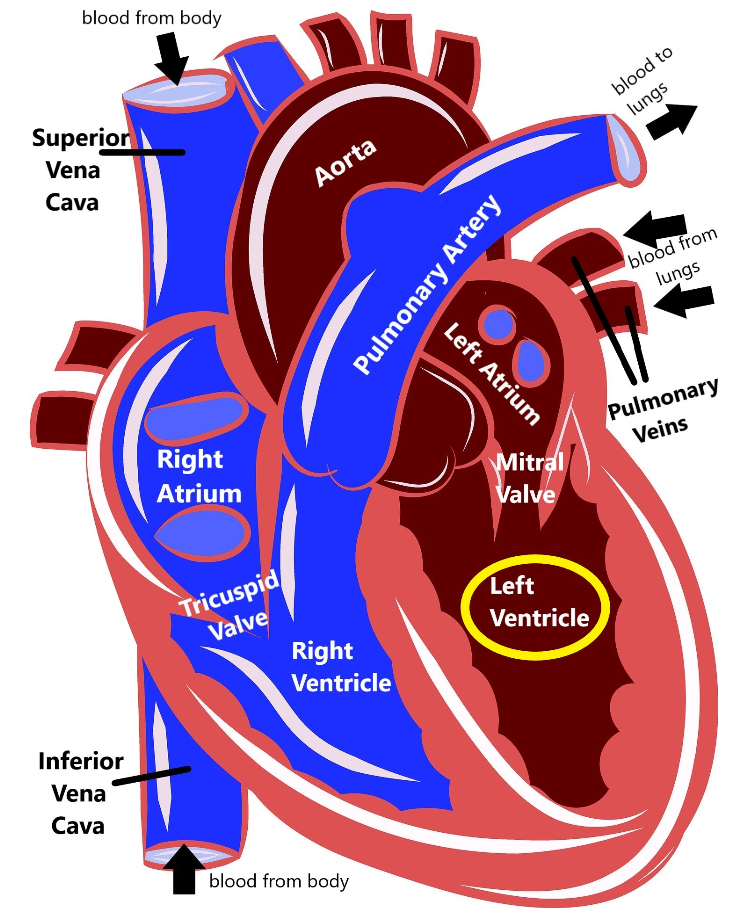
As you can see, the heart is divided into four main chambers: the left atrium and right atrium at the top, and the left ventricle and right ventricle at the bottom. The blue on the diagram is blood that has already delivered oxygen to the body (deoxygenated blood) and is coming back to the heart to get more. The deoxygenated blood comes into the right atrium via two structures called the superior vena cava and the inferior vena cava. Then the right atrium pumps the blood through the tricuspid valve into the right ventricle. The right ventricle then pumps the blood out of the pulmonary artery to the lungs.
When the blood gets to the lungs, it gathers up more oxygen and then is pumped through the pulmonary veins into the left atrium. The left atrium pumps blood through the mitral valve into the left ventricle. Then, the left ventricle pumps blood out the aorta, which delivers it back out to the body. This is how our blood can circulate and repeatedly carry oxygen to all the different organs and tissues in our body.
The heart also has an electrical system that sends out signals that trigger the heart muscles to contract, which controls our heart rate and our heart rhythm. The electrical signal starts at the top of the heart called the sinoatrial node, which functions as a kind of a natural pacemaker, and that signal travels down the heart, stimulating the atria and then the ventricles to contract, which produces our heartbeat.
Cardiovascular Disease
Cardiovascular disease is a term that can be used to describe a group of health conditions that affect the heart and/or blood vessels. There are many different things that can contribute to our cardiovascular system not functioning as it should, including age, environment, and lifestyle factors such as diet and exercise. There are certain types of cardiovascular disease that can be more strongly genetic, or hereditary, and can appear to run in families.
Conditions that directly affect the heart are generally broken up into two main categories: arrhythmias (which affect the electrical system in the heart), and cardiomyopathies (which affect the structure and muscle of the heart). Some heart conditions affect both the rhythm and the structure of the heart, so may fit into both the arrhythmia and cardiomyopathy categories.
Conditions that affect the cardiovascular system as a whole can lead to a buildup of cholesterol in the body or an increased chance for blood vessels to rupture. Other conditions may include those that affect the formation and overall structure of the heart and/or blood vessels, called congenital heart defects.
It can be difficult to determine if someone has an increased hereditary risk for cardiovascular disease, so meeting with a provider who is experienced with cardiovascular genetics is important to get the most up-to-date and accurate information. Below are some cardiovascular conditions that may be more likely to be hereditary:
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a disease that causes the heart muscle to break down, primarily in the right ventricle.
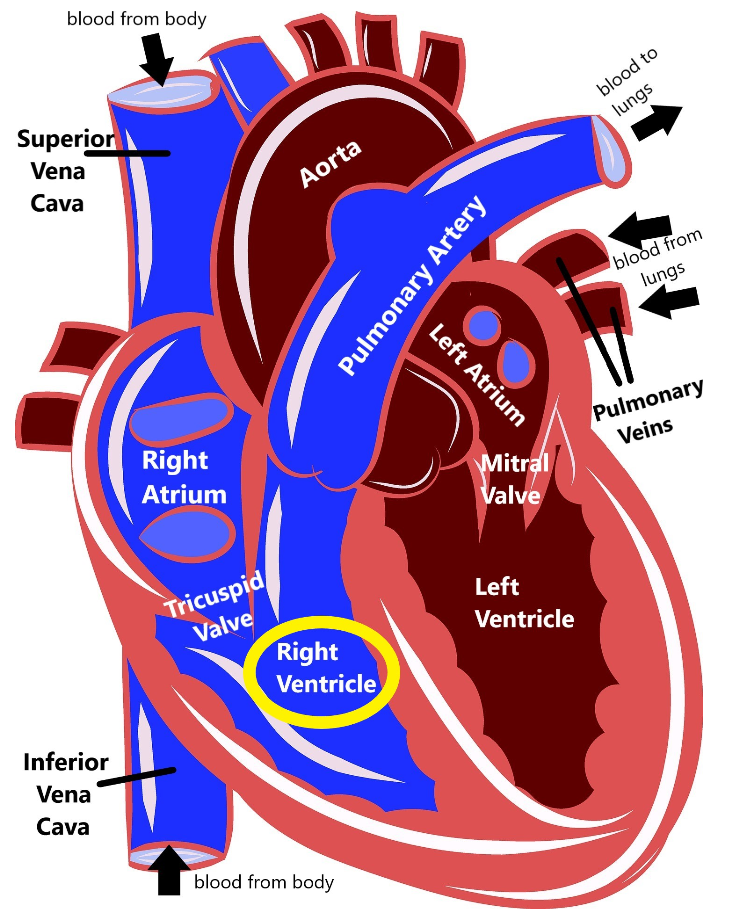
When this muscle breaks down, there is an increased risk that the heart’s electrical system cannot work properly (causing an arrhythmia, or an abnormal heartbeat), which in turn increases the risk for sudden death. Because ARVC affects both the electrical system and the muscle of the heart, it technically fits into both the ‘arrhythmia’ and ‘cardiomyopathy’ categories of heart disease.
People who have ARVC may have no symptoms until adulthood, and the first things many individuals with ARVC will notice are heart palpitations, fainting, and feeling light-headed. People who have more advanced ARVC can also develop edema (swelling of the legs or abdomen) and shortness of breath. Over time if the heart muscle develops significant damage it can cause heart failure.
Some individuals who have ARVC may not have any obvious symptoms, but can still be at a higher risk of sudden death, particularly during strenuous exercise. The average age for someone with ARVC to be diagnosed is 31 years old.
Causes
There are many different genes that have been found to cause different types of ARVC in families, some of these include:
- PKP2 (34-74% of all cases of ARVC)
- DSG2 (5-26% of all cases of ARVC)
- DSP (2-39% of all cases of ARVC)
- DSC2 (1-2% of all cases of ARVC)
- TMEM43 (unknown percentage of all cases of ARVC)
- LMNA (unknown percentage of all cases of ARVC)
ARVC is thought to affect between 1 in 1000 to 1 in 1250 of people. It is possible that it is more common than this, and that people who are affected that haven’t shown any symptoms just don’t know that they have it.
Diagnosing ARVC
While genetic testing for ARVC can be helpful to establish a diagnosis, medical providers may also use other medical tests, such as an ECG or an echocardiogram. Some red flags that can increase the chance for ARVC in the family include:
- Fainting or passing out
- Heart palpitations
- Cardiac arrest (sudden heart failure)
- Abnormal ECG
- Abnormal right ventricle seen through heart imaging, such as an echocardiogram or cardiac MRI
Many families may have these red flags in their family history and DO NOT have ARVC. However, someone with a strong pattern of these or other heart issues may be at a higher chance to have ARVC, and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for ARVC
Treatment for ARVC can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with ARVC. Medications to help prevent arrhythmias and surgically implanting a defibrillator are some common therapies for ARVC. It is also usually recommended that people who have ARVC avoid regular strenuous activity because it can put more strain on the heart muscles.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Brugada syndrome is a genetic condition that interferes with the heart’s normal electrical rhythm (called an arrhythmia) in the lower part of the heart (the ventricles).
Most people with Brugada syndrome do not have symptoms until adulthood, although in rare cases they can occur as early as infancy. These symptoms include fainting, difficulty breathing, seizures, and sudden death, and often happen when the person is resting or asleep.
The risk for sudden death from Brugada syndrome is highest at around age 40, although it is also suspected to play a part in some instances of Sudden Infant Death Syndrome (SIDS), which is a significant cause of death in children under a year old. Brugada syndrome was also at one time called Sudden Unexplained Nocturnal Death Syndrome (SUNDS).
Causes
Brugada syndrome can be inherited/genetic, or can be something that someone develops because of something they were exposed to (acquired). The acquired form of Brugada syndrome has been linked primarily to certain medications, having abnormally high levels of calcium, or having abnormally high or low levels of potassium.
Although there are approximately 23 genes that have been suspected to cause hereditary Brugada syndrome, harmful changes (called pathogenic variants) in the SCN5A gene are the most common cause that we know of thus far (causing 15-30% of all cases of Brugada syndrome).
Brugada syndrome is thought to affect approximately 1 in 2000 (0.05%) people. It is possible that it is more common than this, and that people who are affected that haven’t shown any symptoms just don’t know that they have it. Some studies have estimated that Brugada syndrome is the cause for 4-12% of all unexpected sudden deaths, and for up to 20% of sudden deaths in people who don’t have any obvious heart problems.
Most people with hereditary Brugada syndrome have a parent who also has it, but 1% of people are the first ones in their family to have it (called de novo).
Diagnosing Brugada syndrome
While genetic testing for Brugada syndrome can be helpful to establish a diagnosis, medical providers may also use other medical tests, such as an ECG, an echocardiogram, or your family history. Some red flags in the family history that may increase the chance for Brugada syndrome include people with:
- Repeated fainting or passing out
- Ventricular fibrillation: when the lower chambers of the heart (ventricles) quiver instead of beating like normal
- Self-terminating polymorphic ventricular tachycardia: when the ventricles begin to beat too fast, but then fix themselves
- Cardiac arrest (sudden heart failure)
- Sudden cardiac death
Many families may have these red flags in their family history and DO NOT have Brugada syndrome. However, someone with a strong pattern of these or other heart issues may be at a higher chance to have Brugada syndrome, and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for Brugada syndrome
Treatment for Brugada syndrome can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with Brugada syndrome. Some options for medical management can include medication, regular monitoring with an ECG, and a surgically-implantable defibrillator.
People who have Brugada syndrome should also be careful to avoid certain medications (such as antipsychotic or antidepressant drugs that block sodium, or certain drugs to treat arrhythmias), anesthesia, and high fevers, when possible.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a genetic condition that interferes with the heart’s normal electrical rhythm (called an arrhythmia) in the lower part of the heart (the ventricles).
This interference makes the ventricles contract more quickly than normal, which speeds up the heart rate (tachycardia). Repeated occurrences of tachycardia increase the chance for heart failure (cardiac arrest) and sudden death.
People who have CPVT generally start to see symptoms between the ages of 7-12 years old. Symptoms of CPVT can include fainting, lightheadedness, dizziness, and heart attack. Strenuous physical activity and emotional stress can increase the chance for someone with CPVT to show symptoms.
Causes
Approximately half of all cases of CPVT can be linked to the RYR2 gene, and about 1-2% of all cases are linked to the CASQ2 gene. The remaining individuals may have a genetic link to their CPVT, but it is in a gene that we do not yet know about yet.
CPVT is thought to affect approximately 1 in 10,000 people. It is possible that it is more common than this, and that people who are affected that haven’t shown any symptoms just don’t know that they have it.
Diagnosing CPVT
While genetic testing for CPVT can be helpful to establish a diagnosis, medical providers may also use other medical tests, such as an ECG, or an echocardiogram. Some red flags that can increase the chance for CPVT in the family include:
- Fainting or passing out during physical activity or stressful emotional periods before the age of 40
- History of dizziness or a rapid, irregular heart beat (palpitations) during exercise or periods of emotional stress
- Emotional stress or strenuous activity that triggers sudden unexpected cardiac death
- Ventricular arrhythmias (abnormal heart beat) during exercise
- Ventricular fibrillation (when the lower chambers of the heart quiver instead of beating like normal) during times of acute stress
- A structurally normal heart
Many families may have these red flags in their family history and DO NOT have CPVT. However, someone with a strong pattern of these or other heart issues may be at a higher chance to have CPVT, and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for CPVT
Treatment for CVPT can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with CVPT. Some options for medical management can include medication, regular monitoring with an ECG, and a surgically-implantable defibrillator.
People who have CVPT should also be careful to avoid competitive sports or other strenuous exercises, as well as significant emotional stress (when possible).
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Long QT syndrome (LQTS) is a disorder that can cause erratic and irregular heartbeat. Our heart is controlled by an electrical system that helps determine how fast our heart should beat (your heart rate). It does this by using electrical signals to coordinate the heart muscles to beat together so the heart can pump blood efficiently. If this electrical system does not work properly, then it can cause the heart to beat too fast or too slow, or stop the heart from beating altogether. This electrical signal starts at a part of the heart called the sinoatrial node, which is located in the right atrium of heart.
This electrical signal works its way down the heart to signal the muscles to contract together, which lets our heart more efficiently pump blood to the rest of our bodies. A medical device called an echocardiogram can measure this electrical pulse by looking five distinct waves, called P, Q, R, S, and T. The electrical activity in the bottom two chambers of the heart (the ventricles) is measured by looking at the length between the Q and T waves (called the QT interval).
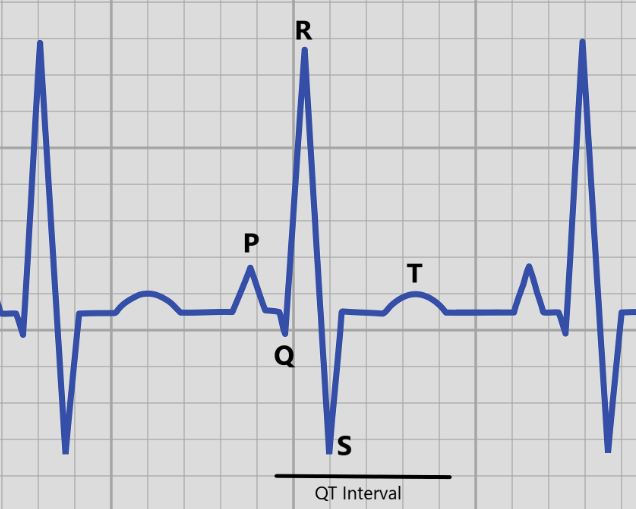
If someone has a QT interval that is too long, that can lead to a longer time in between heartbeats (which explains the name Long QT syndrome). This abnormal heartbeat, called an arrhythmia, can lead to dizziness, lightheadedness, and fainting, or in more serious cases seizures or sudden cardiac death. Some people with LQTS may not have any obvious symptoms, but can still be at a higher risk of sudden death. Two people with LQTS, even within the same family, can have very different signs and symptoms.
Sudden cardiac death can happen anytime from infancy to advanced adulthood, but is more common in the teens and 20s. For about 10-15% of people who have LQTS, sudden death will be the first symptom they have.
Causes and subtypes
There are 15 different subtypes of LQTS that are all caused by different genes. The three most common subtypes make up about 75% of cases of LQTS:
- KCNQ1: type 1 (30-35% of all cases of LQTS)
- KCNH2: type 2 (25-30% of all cases of LQTS)
- SCN5A: type 3 (5-10% of all cases of LQTS)
The risk of sudden death is the same for all three of the common subtypes of LQTS (6-8%). However, the chance to have another symptoms (such as lightheadedness or fainting) is quite different (63% for type 1, 46% for type 2, and 18% for type 3). That means that 37% of people with LQTS type 1, 54% of people with LQTS type 2, and 82% of people with LQTS type 3 will never show any symptoms.
There are also different things that can trigger these health concerns depending on what type of LQTS someone has. Periods of intense emotion or strenuous exercise (particularly swimming) can trigger a heart issue in people with type 1, which people with type 2 can be triggered by emotion (such as excitement or fear), exercise, or during their sleep. Most people with LQTS type 3 will experience related heart issues during their sleep.
The other known 12 subtypes are more rare, and each account for less than 1% of all cases of LQTS. Approximately 20% of families who have LQTS do not have a genetic cause that can be found, which means there are likely other genes that we don’t know about yet that cause LQTS in some families. A small number of cases of LQTS can be acquired, or caused by things like medications or other underlying health issues, rather than genetic.
Approximately 1 in 2000 to 1 in 2500 people are thought to have LQTS. It is possible that it is more common than this, and that people who are affected that haven’t shown any symptoms just don’t know that they have it.
Diagnosing LQTS
While genetic testing for LQTS can be helpful to establish a diagnosis, medical providers may also use other medical tests, such as an ECG and your family history. Some red flags that can increase the chance for LQTS in the family include:
- A history of fainting
- A history of a child or young adult that died suddenly
- A history of arrhythmia
Many families may have these red flags in their family history and DO NOT have LQTS. However, someone with a strong pattern of these or other heart issues may be at a higher chance to have LQTS, and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for LQTS
Treatment for LQTS can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with LQTS. This treatment can include medications (including beta blockers and sodium channel blockers) or surgically implanting a defibrillator.
Medical management is also different depending on what type of LQTS someone has. For example, because people with type 1 are more likely to have a heart issue during activity strenuous activity is not recommended, while people with type 2 should try to avoid loud noises because those startling noises can trigger a heart issue.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Familial dilated cardiomyopathy (DCM) is a health condition that can run in families that impacts the heart’s ability to function how it should due to the heart muscle in the left ventricle becoming thin and weak. Because the muscle becomes more thin, the space inside the left ventricle, called the chamber, gets bigger (dilated).
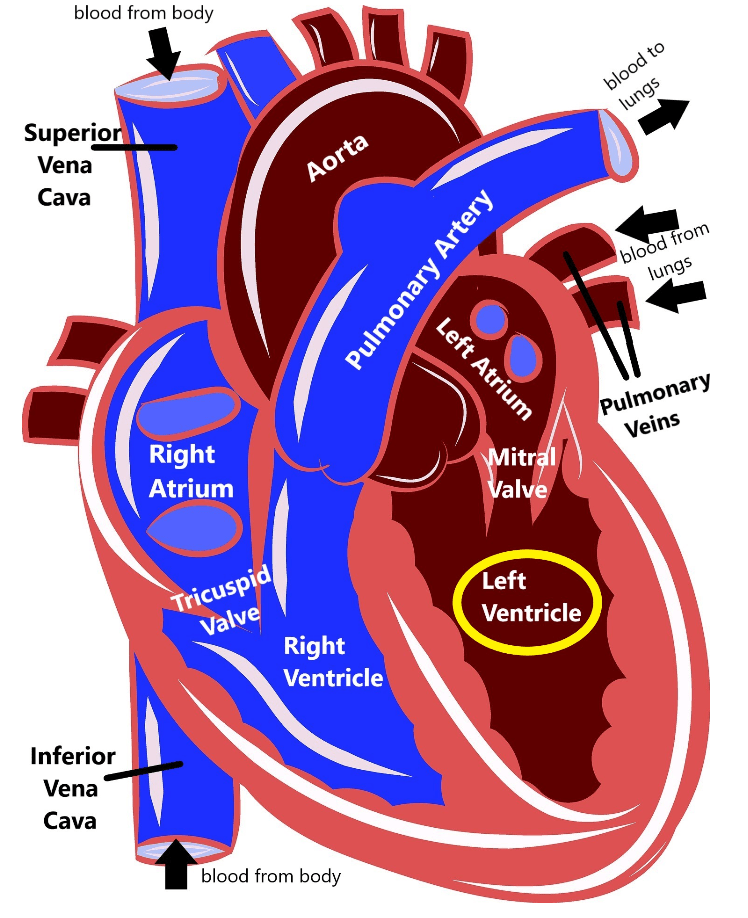
When the space inside the heart is bigger and the heart muscle is weak, the heart can’t pump blood as efficiently as it should. This means that the heart has to work harder to try to pump a normal amount of blood, which further causes the heart muscle to thin and grow weaker. Eventually, DCM can lead to heart failure.
Cardiomyopathies (diseases of the heart muscle) can come in several forms, but DCM is the most common. It typically affects people who are between the ages of 20-60 years old, and can also lead to arrhythmias (irregular heartbeat), blood clots, or sudden death. Some studies have estimated that approximately 1 in 250 people will have DCM, and males are three times more likely to have DCM than women.
Causes
About half of all cases of DCM are inherited, or genetic, and can be passed down in families. The other half are either acquired through environmental causes (such as exposures or medications), or the cause is unknown. Approximately 30 genes have thus far been discovered that can cause DCM in families, including:
- TTN (10-20% of all DCM)
- LMNA (6% of all DCM)
- MYH7 (4.2% of all DCM)
- MYH6 (3-4% of all DCM)
- MYBPC3 (2-4% of all DCM)
- SCN5A (2-4% of all DCM)
- TNNT2 (2.9% of all DCM)
- BAG3 (2.5% of all DCM)
- ANKRD1 (2.2% of all DCM)
- TPM1 (unknown percentage of all DCM)
- TNNI3 (unknown percentage of all DCM)
- ACTC1 (unknown percentage of all DCM)
- DSG2 (unknown percentage of all DCM)
Diagnosing Dilated Cardiomyopathy
While genetic testing for DCM can be helpful to establish a diagnosis or to determine what form of DCM someone has, a medical provider who has experience with DCM can usually diagnose it by using other tests. An echocardiogram or cardiac MRI can look to see if the left ventricle is measuring larger than expected, as well as how much blood the left ventricle is pumping (called systolic function). If the left ventricle is enlarged AND someone’s systolic function is less than 50% of what it should be, that would lead to a clinical diagnosis of DCM.
People who have DCM usually start to show symptoms in their 40s, 50s, or 60s, although symptoms can begin at any age.
Medical Management for Dilated Cardiomyopathy
Treatment for DCM can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with DCM. Some options for medical management can include medication, regular cardiac monitoring, a surgically-implantable defibrillator, and a heart transplant. People who have DCM should try to avoid things like strenuous physical activity and excessive alcohol consumption.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Familial hypertrophic cardiomyopathy (FHCM) is a disease that causes the muscle of the heart to become thicker than normal (called hypertrophy). There are several parts of the heart where the muscle can hypertrophy, but it most frequently happens in the muscle that separates the two bottom chambers of the heart (the ventricles).
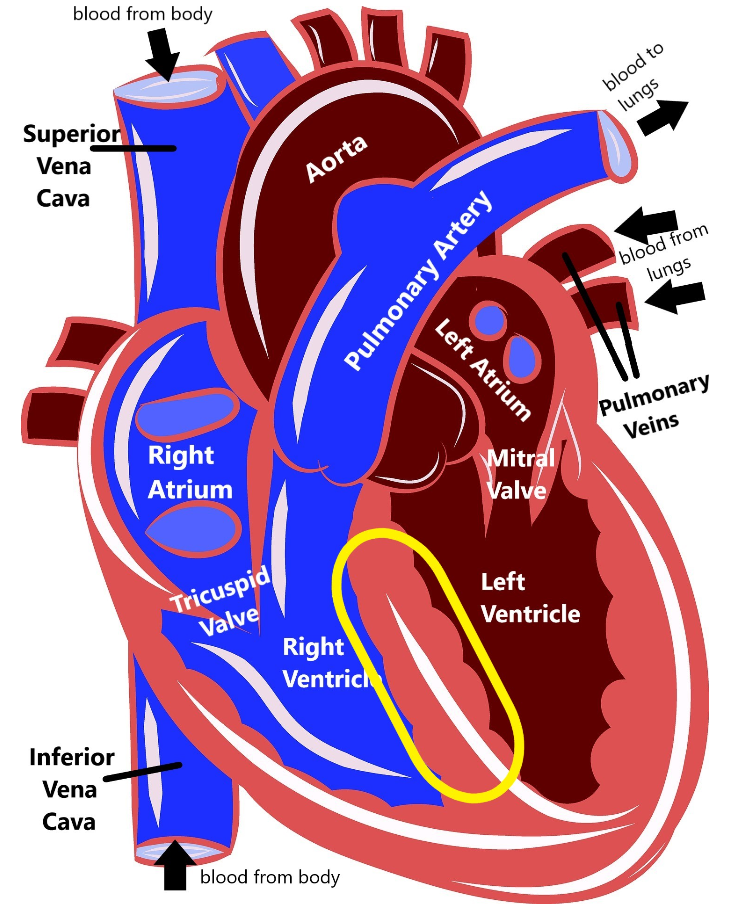
When this muscle becomes more thick, it can lead to the heart not being able to pump oxygenated blood around the body. Sometimes this thickened muscle can block the flow of blood out of the heart, which can lead to a heart murmur, or can disrupt the heart’s electrical system, causing an arrhythmia. People who have FHCM are more likely to start showing symptoms in their teens to young adulthood, but some people do not have any health issues until later in life.
The specific health issues that affect someone with FHCM can be very different from person to person, even with people in the same family. Some people may begin to notice feeling dizzy or lightheaded, fainting, having chest pain or shortness of breath, or heart palpitations, while other people will not have any symptoms. However, a major concern for individuals with FHCM is the risk for sudden death, even if they have had no prior symptoms of heart trouble.
Causes
There are over a dozen different genes that have found to cause FHCM:
- MYH7: 40% of all FHCM
- MYBPC3: 40% of all FHCM
- TNNT2: 5% of all FHCM
- TNNI3: 5% of all FHCM
- TPM1: 2% of all FHCM
- MYL3: 1% of all FHCM
- PRKAG2: unknown percentage of all FHCM
- MYL2: unknown percentage of all FHCM
- ACTC1: unknown percentage of all FHCM
It is estimated that approximately 1 in 500 people have FHCM. Most (~70%) people with FHCM will have a parent who also has FHCM. For the other 30%, they will be the first ones in their family to have FHCM (called de novo).
Diagnosing FHCM
While genetic testing for FHCM can be helpful to establish a diagnosis, medical providers may also use other medical tests, such as an echocardiogram, a cardiac MRI, or your family history. Red flags in the family history that may increase the chance for FHCM include people with:
- Heart failure
- Hypertrophic cardiomyopathy
- Heart transplants
- Unexplained or sudden death, particularly before the age of 40
- Arrhythmias
- Unexplained stroke or blood clotting disease
Many families may have these red flags in their family history and DO NOT have FHCM. However, someone with a strong pattern of these or other heart issues may be at a higher chance to have FHCM, and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for FHCM
Treatment for FHCM can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with FHCM. Medications, certain therapies, and implantable devices, such as a pacemaker or implantable cardiac defibrillator may be part of the treatment plan. It is also recommended that people who are known to have FHCM avoid high intensity exercise (such as endurance training or running sprints), heavy weight lifting, dehydration, and certain medications (including some blood pressure medications).
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
The muscles in our heart are usually smooth and firm, which is how they most efficiently pump blood around our bodies. Left ventricular noncompaction (LVNC) is a heart condition that causes the muscle of one of the bottom chambers of the heart (the left ventricle) to not develop correctly.
Instead of having smooth and firm muscle, the left ventricle in people with LVNC has muscle that is spongy, weak, and thick. This muscle cannot contract and relax how heart muscles normally should, so it is not able to pump blood as well out of the left ventricle into the rest of the body.
Some people who have LVNC may experience signs and symptoms, including:
- Blood clots
- A heart arrhythmia (abnormal heartbeat)
- Heart palpitations
- Extreme fatigue, particularly during exercise
- Shortness of breath, dizziness, and fainting
- Lymphedema
Other people who have LVNC may have no symptoms at all, but could be at a higher risk for sudden cardiac death. Although some individuals with LVNC may not have significant health issues, about two-thirds will go on to develop heart failure.
Causes
Only about 20-40% of cases of LVNC have a underlying genetic cause that can currently be found. Some cases of LVNC may be the result of other underlying health conditions (such as some metabolic or mitochondrial genetic disorders), but a cause cannot be identified for many people with LVNC.
There are likely many genes that can cause or increase the chance for someone to develop LVNC, but the ones that are currently most commonly tested for are MYH7, MYBPC3, SCN5A, TPM1, ACTC1, LMNA, and TNNT2.
It is estimated that approximately 8 to 12 of every one million people have LVNC. It is possible that it is more common than this, and that people who are affected that haven’t shown any symptoms just don’t know that they have it.
Diagnosing LVNC
While genetic testing for LVNC can be helpful to establish a diagnosis, medical providers may also use other medical tests, such as an echocardiogram or your family history. Red flags in the family history that may increase the chance for LVNC include people with:
- cardiomyopathies
- arrhythmias
- sudden or unexplained death
- feeding issues in babies and young children
- extreme fatigue during exercise (exercise intolerance)
Many families may have these red flags in their family history and DO NOT have LVNC. However, someone with a strong pattern of these or other heart issues may be at a higher chance to have LVNC, and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for LVNC
Treatment for LVNC can sometimes vary depending on the individual person and their specific health concerns. Certain medications, such as aspirin to help prevent blood clots, and implantable devices, such as a pacemaker or implantable cardiac defibrillator may be part of the treatment plan. Some patients with LVNC may need a heart transplant. It also may be recommended that people with LVNC avoid high-intensity or endurance sports, and to monitor their breathing and energy levels during routine exercise.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a disease that causes the heart muscle to break down, primarily in the right ventricle.
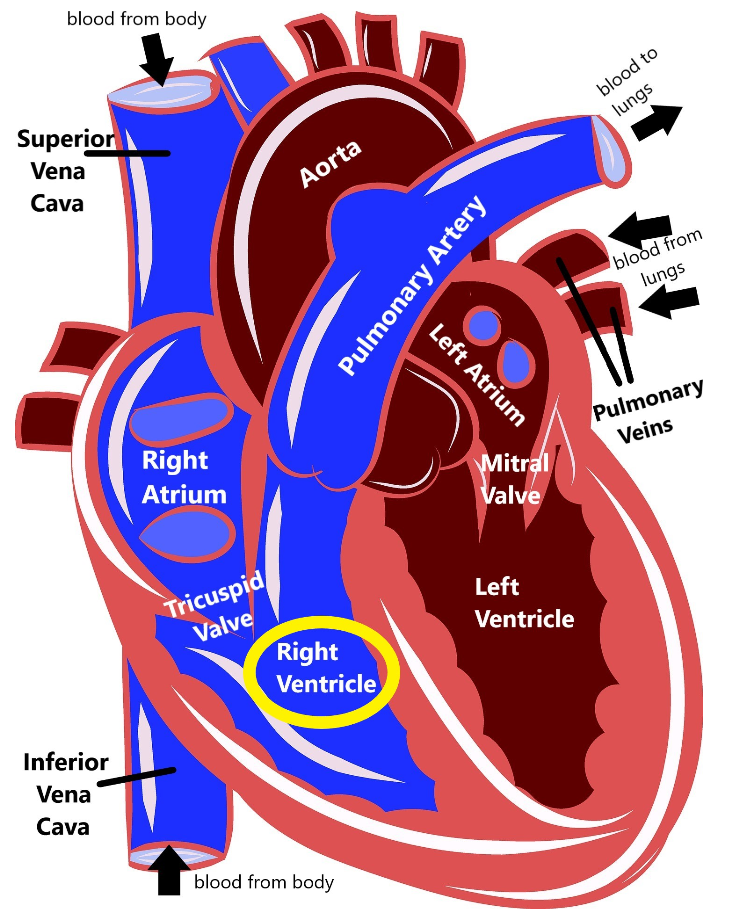
When this muscle breaks down, there is an increased risk that the heart’s electrical system cannot work properly (causing an arrhythmia, or an abnormal heartbeat), which in turn increases the risk for sudden death. Because ARVC affects both the electrical system and the muscle of the heart, it technically fits into both the ‘arrhythmia’ and ‘cardiomyopathy’ categories of heart disease.
People who have ARVC may have no symptoms until adulthood, and the first things many individuals with ARVC will notice are heart palpitations, fainting, and feeling light-headed. People who have more advanced ARVC can also develop edema (swelling of the legs or abdomen) and shortness of breath. Over time if the heart muscle develops significant damage it can cause heart failure.
Some individuals who have ARVC may not have any obvious symptoms, but can still be at a higher risk of sudden death, particularly during strenuous exercise. The average age for someone with ARVC to be diagnosed is 31 years old.
Causes
There are many different genes that have been found to cause different types of ARVC in families, some of these include:
- PKP2 (34-74% of all cases of ARVC)
- DSG2 (5-26% of all cases of ARVC)
- DSP (2-39% of all cases of ARVC)
- DSC2 (1-2% of all cases of ARVC)
- TMEM43 (unknown percentage of all cases of ARVC)
- LMNA (unknown percentage of all cases of ARVC)
ARVC is thought to affect between 1 in 1000 to 1 in 1250 of people. It is possible that it is more common than this, and that people who are affected that haven’t shown any symptoms just don’t know that they have it.
Diagnosing ARVC
While genetic testing for ARVC can be helpful to establish a diagnosis, medical providers may also use other medical tests, such as an ECG or an echocardiogram. Some red flags that can increase the chance for ARVC in the family include:
- Fainting or passing out
- Heart palpitations
- Cardiac arrest (sudden heart failure)
- Abnormal ECG
- Abnormal right ventricle seen through heart imaging, such as an echocardiogram or cardiac MRI
Many families may have these red flags in their family history and DO NOT have ARVC. However, someone with a strong pattern of these or other heart issues may be at a higher chance to have ARVC, and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for ARVC
Treatment for ARVC can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with ARVC. Medications to help prevent arrhythmias and surgically implanting a defibrillator are some common therapies for ARVC. It is also usually recommended that people who have ARVC avoid regular strenuous activity because it can put more strain on the heart muscles.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Familial hypercholesterolemia (FH) is an inherited condition where the body is not able to get rid of extra cholesterol, causing it to build up in the bloodstream and other parts of the body. This buildup of cholesterol can narrow the heart valves and arteries, which increases the chance for cardiovascular disease such as stroke, heart attack, and heart disease. These conditions are also often diagnosed at younger ages in people with FH than would normally be expected. As many as 30-50% of people with FH may also develop small yellowish patches of cholesterol buildup called xanthomas. Xanthomas usually occur around the eyelids and in the tendons in the feet, hands, knees, and elbows, and may get smaller with treatment. About half of all men with FH will have a health issue with their heart or their arteries by the age of 50, while 85% will have a heart attack by the age of 60. In women with FH, about 30% will experience a health issue with their heart or arteries by the age of 60.
Cholesterol is needed by the body to work properly, and it helps the body to make hormones, vitamin D, and certain substances that help with food digestion. There are two main types of cholesterol, called HDL and LDL. LDL works to deliver cholesterol to your arteries, while HDL works to move cholesterol to your liver. The liver removes extra cholesterol from the body, preventing it from building up. LDL cholesterol is the type that most doctors are talking about when they tell someone they have high levels of the ‘bad’ cholesterol.
For most people with high LDL cholesterol, their diet or lifestyle causes an increase in the amount of LDL cholesterol in their body, and the liver can not get rid of the extra cholesterol fast enough. For people who have FH, their high levels of LDL cholesterol are not directly because of their diet or lifestyle (although those also do play an important part), but because their liver is not able to remove the extra LDL cholesterol.
Causes
Approximately 70-95% of cases of FH are caused by harmful changes (called pathogenic variants) in one of three genes: LDLR (60-80% of all cases of FH), APOB (1-5% of all cases of FH), and PCSK9 (0-3% of all cases of FH). The remaining cases have an unknown cause, but may be due to pathogenic variants in other genes that we do not know about yet.
Recent studies have estimated that as many as 1 in 200 (0.5%) people may have FH, and that 2-3% of all heart attacks that happen before the age of 60 may be due to FH. Between 1 in 160,000 and 1 in 250,000 people inherit a FH gene from each parent that has a pathogenic variant in it (called homozygous). Most people who have two non working FH genes have severe coronary artery disease by their mid-20s, and the risk for sudden death or bypass surgery is high during the teenage years. People who are homozygous may also develop xanthomas in between their fingers.
Diagnosing FH
While genetic testing for FH can be helpful to establish a diagnosis, medical providers may use other pieces of information from lab tests, a physical exam, and the family history to determine if someone is at a higher chance to have FH. Some red flags that can increase the chance for FH in a family include:
- Extremely high cholesterol
- In adults, this would be an LDL cholesterol level of over 190 mg/dL, or a total cholesterol level of over 310 mg/dL.
- In children, this would be an LDL cholesterol level of over 130 mg/dL, or a total cholesterol level of over 230 mg/dL.
- A personal or family history of early-onset (generally thought to be before the age of 45 in men and before the age of 50 in women) coronary artery disease or cardiovascular disease (including angina, heart attack, and vascular disease)
- Physical exam that finds xanthomas at any age or corneal arcus (a gray or white arc or ring around the colored part of the eye) before the age of 45
It is thought that most people (up to 90%) who have FH have not been properly diagnosed. If you or members of your family have any history that is concerning for the signs and symptoms of FH, you should speak with a specialist, such as a cardiovascular specialist or genetic counselor, to get more information.
Medical Management for FH
Treatment for FH can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with FH. Some options for medical management can include regular blood work, medications, dietary recommendations (reduced saturated fat and increased soluble fiber), and imaging (including echocardiograms and CT angiograms).
People with FH should avoid risk factors for cardiovascular and coronary artery disease, such as smoking, high fat and high cholesterol diet, obesity, and high blood pressure. Regular physical activity and following dietary recommendations can help with weight control as well as overall cardiovascular health.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Familial thoracic aortic aneurysm and dissection (familial TAAD) is a hereditary condition that increases the chance for a specific type of heart issue. The aorta is the upper part of the heart that pumps oxygenated blood from our heart out to the rest of our body:
People who have familial TAAD can develop a weakened aorta. This weakening in the wall of the aorta can lead to pocket that bulges out of the side of the aorta, called an aneurysm. There are usually no physical signs or symptoms of an aneurysm, but some people can see changes in their breathing (shortness of breath, hoarseness, or wheezing), pain or swelling in the head, neck or upper body, or sudden sharp pain in the chest or back.
The weakening of the aortic wall can also lead to a widening of the aorta, called aortic dilation. This bulging and weakening of the wall of the aorta can also lead to an aortic dissection, or when the aorta ruptures. An aortic dissection can lead to death because it causes blood to flow outside of the aorta, and not enough blood is being pumped out to the rest of the body.
Individuals who have familial TAAD most frequently will have aortic dilation as the first sign, but some people may have a dissection without having any other previous symptoms. Familial TAAD may be an isolated hereditary health condition, or it could be associated to other underlying genetic conditions like Marfan syndrome, Loeys-Dietz syndrome, and vascular Ehlers-Danlos syndrome.
Causes
At least 20% of all aortic aneurysms and dissection are thought to be due to familial TAAD. That means the remaining 80% are likely not strongly genetic, and are multifactorial, or are due to genetic causes that we do not yet know about.
There are over a dozen genes that have been found to cause familial TAAD:
- ACTA2 (12-21% of all cases of familial TAAD)
- TGFBR2 (5% of all cases of familial TAAD)
- FBN1 (3% of all cases of familial TAAD), which also causes Marfan syndrome
- TGFBR1 (3% of all cases of familial TAAD)
- SMAD3 (2% of all cases of familial TAAD)
- LOX (1.5% of all cases of familial TAAD)
- FOXE3 (1.4% of all cases of familial TAAD)
- MAT2A (1% of all cases of familial TAAD)
- MYH11 (1% of all cases of familial TAAD)
- MYLK (1% of all cases of familial TAAD)
- PRKG1 (1% of all cases of familial TAAD)
- TGFB2 (1% of all cases of familial TAAD)
- MFAP5 (0.25% of all cases of familial TAAD)
- BGN (rare cause of familial TAAD)
- COL3A1 (rare cause of familial TAAD), which also causes vascular Ehlers-Danlos syndrome
- TGFB3 (rare cause of familial TAAD)
Diagnosing familial TAAD
While genetic testing for familial TAAD can be helpful to establish a diagnosis, medical providers may use other pieces of information from other lab tests or someone’s medical and family history to make the diagnosis. Some these tests may include an echocardiogram, a cardiac CT scan, or a cardiac MRI. A family history of aortic or general cardiovascular disease could increase the chance for familial TAAD in a family. Many families will have a history of heart disease and DO NOT have familial TAAD, however someone with a strong pattern of these heart issues may be at a higher chance to have familial TAAD and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for familial TAAD
Treatment for familial TAAD can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a provider who is familiar with familial TAAD. Medical management for more specific forms of familial TAAD, such as Marfan syndrome, Loeys-Dietz syndrome, and vascular Ehlers-Danlos syndrome, may also include screening for other related health concerns. Some options for medical management include an echocardiogram, a cardiac CT scan, or a cardiac MRI in order to look at the aorta to assess for dilation or aneurysm, medications called beta blockers that help to lower the blood pressure (which reduces the stress on the heart), and avoiding contact sports. Other risk factors for heart disease, such as smoking and high cholesterol, should also be avoided when possible.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Genetics & Connective Tissue Disorders
Our bodies are made up of many different muscles, organs, and bones. Connective tissue is a type of tissue in the body that helps to hold everything together, like a glue for your body. We have over 20,000 different genes in the body. These genes are like instruction manuals for how to build a protein, and each protein has an important function that helps to keep our body working how it should. There are many of these proteins that either help to make connective tissue, or help the connective tissue function properly. If someone has a non-working copy of one of these genes, it can cause a connective tissue disorder.
There are thought to be over 200 different disorders that can affect our connective tissue, and they are all quite variable. Some can cause very little health impact, while others can cause serious (sometimes even life-limiting) medical concerns.
As genetic research continues to learn more about our genes and how they affect our bodies, there are likely to be more specific types of connective tissue disorders that are discovered. Some of the more common connective tissue disorders we know about thus far include:
Hypermobile Ehlers-Danlos syndrome (hEDS) (previously called EDS, hypermobile type or Type III EDS) is a genetic condition that affects connective tissue. Connective tissue is a type of tissue that helps to hold everything together, like a glue for your body. hEDS is characterized by generalized joint hypermobility and associated complications such as joint pain and dislocations. People who have hEDS may also have:
- soft/velvety skin
- unexplained stretch marks (also called striae)
- easy bruising
- recurrent hernias
- osteoarthritis
- chronic pain
- chronic fatigue
- mild aortic root dilation (stretching of the main blood vessel that carries blood from the heart to the body)
- mitral valve prolapse
- symptoms related to dysautonomia (such as neurally mediated hypotension (NMH) or postural orthostatic
- tachycardia syndrome (POTS))
- varying gastrointestinal/digestive difficulties
In general, people who have hEDS have skin that may appear more transparent, hyperextensible (stretchy), and fragile than normal. There may also be some abnormal scarring, but this typically less common than in other types of Ehlers-Danlos syndrome. Other forms of EDS can also have issues with the soft tissue around their internal organs, but that is not usually seen with hEDS.
Individuals who have hEDS may notice a progression of some physical symptoms as they get older. What can start out as being extra flexible or hypermobile often leads to decreased mobility along with pain, fatigue, and other related complications.
hEDS can range in severity, and there can be variability within a family. Individuals may have few or no symptoms while others may have more severe and early-onset health issues.
Causes
The exact underlying genetic cause for many individuals with hEDS is unknown. Most cases of hEDS appear to follow an autosomal dominant inheritance pattern, meaning that any children of someone who has hEDS also have a 50% chance to have hEDS. This also means that most people with hEDS have a parent that is also affected. hEDS has traditionally been under-diagnosed, so it is very possible that there could be other members of a family who have hEDS that have just not been diagnosed. Other factors (such as gender, age, activities) may also impact whether someone with hEDS has symptoms and how severe those symptoms are.
Some individuals with hEDS have been reported to have pathogenic variants in the TNXB gene. However, this is rare and it is unknown how pathogenic variants in this gene lead to the symptoms seen in hEDS. It is likely that there are other genes that contribute to hEDS in some families that have just not yet been discovered. hEDS is thought to affect approximately 1 in 5000 to 1 in 20,000. Because the signs and symptoms in individuals with hEDS are variable, it is possible that hEDS is much more common but that it is just not being diagnosed.
Diagnosing Hypermobile Ehlers-Danlos syndrome
Genetic testing is currently very rarely helpful to establish a diagnosis of hEDS, so medical providers use other pieces of information, such as a physical exam and family history, to help establish the diagnosis. There are two different sets of criteria that currently exist for medical providers to evaluate whether or not someone has hEDS: the Villefranche criteria for EDS hypermobile type, and the Beighton criteria for joint hypermobility syndrome (JHS). There is much overlap between the Villefranche and Beighton criteria, and what age you are when the evaluation takes place may determine if you meet the criteria or not.
In 2017 the International EDS Consortium published criteria for the diagnosis for hEDS. For someone to have a diagnosis of hEDS, they need to have:
- Generalized joint hypermobility
- At least two of the following:
- Other signs or symptoms of a connective tissue disorder (soft or velvety skin, stretchy skin, dilation of the aorta, etc)
- A positive family history of one or more first-degree relatives (parents, siblings, children) that meet the current criteria for hEDS
- Musculoskeletal (the muscles and skeleton together) concerns (chronic limb pain, chronic widespread pain, frequent joint dislocations without trauma)
- Exclusion of other connective tissue disorders
This complex assessment can generally be made by a medical provider that is familiar with hEDS.
Medical Management for Hypermobile Ehlers-Danlos syndrome
Treatment for hEDS can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with hEDS. Medical management can include physical therapy, pain management through exercises or medication, and vitamins or supplements to help strengthen the bones. It is also usually recommended that individuals with hEDS avoid high-impact activity that can increase the risk for dislocations, chronic pain, and osteoarthritis. Imaging of the heart, called an echocardiogram may also be recommended if there is suspicion of dilation of the aorta, and individuals with hEDS may also see a gastroenterologist if they are experiencing digestive issues.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
The vascular form of Ehlers-Danlos syndrome (vEDS) is a connective tissue disorder than can affect many different parts of the body, including the skin, arteries, muscles, and organs. Connective tissue is a type of tissue in the body that helps to hold everything together, like a glue for your body.
People with vEDS can have some or many signs and symptoms, including skin issues (thin, translucent skin that shows signs of premature aging), easy bruising, hypermobile (extra flexible) fingers and toes, joint dislocations, collapsed lung, receding gums, a higher risk for an aneurysm, and a characteristic facial appearance (that can include thin lips, a small chin, a thin nose, and large eyes). Not all people with vEDS will have all of these signs, and two people with vEDS may have very different symptoms, even within the same family. Individuals with vEDS also have a higher chance for their internal organs (particularly the uterus during pregnancy), arteries, and intestines to rupture due to the connective tissue not holding them together as well as it should. Babies who have vEDS have a higher chance to be born with clubfoot and congenital (from birth) hip dislocation.
Causes
Over 95% of cases of vEDS can be linked to pathogenic variants in the COL3A1 gene, which are inherited in an autosomal dominant pattern. This means that a single copy of the pathogenic variant is enough to cause an individual to develop the condition, and anyone who carries the pathogenic variant has a 50% chance to pass it down to any children they have. The remaining individuals may have a genetic link to their vEDS, but it is in a gene that we do not yet know about. vEDS affects approximately 1 in 50,000 to 1 in 200,000 people. Because of the variability of vEDS and the thought that it is very rare, it is likely under-diagnosed in families that have non-vascular health issues. About 50% of people who have vEDS due to the COL3A1 gene have a parent who also has it. The other 50% are the first ones in their family to have it (called de novo).
Diagnosing vEDS
While genetic testing for vEDS can be helpful to establish a diagnosis, medical providers may use other pieces of information from other lab tests or someone’s medical and family history to make the diagnosis. Some red flags that can increase the chance for vEDS in a family include:
- Aneurysms (a bulging out on a blood vessel) that can rupture
- Rupture of the intestines
- Rupture of the uterus during pregnancy
- Other people in the family with a diagnosis or suspicion of vEDS
- Hypermobile joints/frequent joint dislocations
- Thin skin that bruises easily
- Collapsed lung
- Early-onset varicose veins
Many families may have these red flags in their family history and DO NOT have vEDS. However, someone with a strong pattern of these or other heart issues may be at a higher chance to have vEDS, and may benefit from talking about it more with a specialist, such as a cardiologist or a genetic counselor.
Medical Management for vEDS
Treatment for vEDS can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with vEDS. Some options for medical management can include using imaging, such as ultrasound and magnetic resonance angiography, to try to see all of the arteries in the body to look for aneurysms or other abnormalities, and surgery can sometimes be required immediately if there is a rupture of an internal organ, artery, or intestines. Because of this increased risk for random rupture, people with vEDS should seek immediate medical attention for any sudden unexplained pain.
People with vEDS should try to avoid high-contact sports (such as football and rugby), as well as heavy weight lifting or weight training. Because of the increased risk for related complications, it may also be recommended that individuals who have vEDS avoid routine colonoscopy screening as well as any surgeries that are not absolutely necessary. It is important for people with vEDS to monitor their blood pressure. If their blood pressure is high, that can increase the chance for the veins to rupture, so if high blood pressure is found early treatment can be helpful to reduce the risk for complications.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Classical Ehlers-Danlos syndrome (cEDS), previously called EDS-Classic type or Type I and II EDS, is a connective tissue disorder than can affect many different parts of the body, including the skin, arteries, muscles, and organs. Connective tissue is a type of tissue that helps to hold everything together, like a glue for your body.
People with cEDS can have some or many signs and symptoms, including:
- skin hyperextensibility
- smooth, velvety skin
- atrophic scarring
- splitting of the skin following relatively minor trauma
- delayed wound healing
- easy bruising
- hypermobility and complications of joint hypermobility (e.g., sprains, dislocations/subluxations)
Some individuals with cEDS may also experience low muscle tone (hypotonia), and some children show delayed gross motor development. Cardiac and vascular issues are less common, but there may be an increased risk for dilation of the aorta (stretching of the main blood vessel that carries blood from the heart to the body) and spontaneous rupture of the larger arteries.
Causes
More than 90% of cEDS is due to a pathogenic variants in the COL5A1 gene (75-80%) or the COL5A2 gene (10-15%). In more rare cases, classic EDS will be caused by a pathogenic variant in the COL1A1 gene (<1%), which is also associated with another form of EDS (EDS type VII, or the arthrochalasia type). Pathogenic variants in the genes that cause cEDS are inherited in an autosomal dominant pattern, meaning that anyone who carries the pathogenic variant has a 50% chance to pass it down to any children they have. About 50% of individuals with cEDS will have a positive family history and will have inherited it from an affected parent. The remaining 50% of people with cEDS that have no family history are the result of a new (de novo) variant and will be the first ones in their family to have it.
cEDS is thought to affect approximately 1 in 20,000 people, but it is likely that there are affected individuals who have more mild symptoms and have not been diagnosed so it could be more common that current estimates show.
Diagnosing Classical Ehlers-Danlos syndrome
While genetic testing for cEDS can be helpful, medical providers may use other pieces of information, such as a physical exam and family history, to help establish a diagnosis.
The diagnosis of cEDS is often made by a medical provider who is familiar with cEDS who can look for the signs on a physical examination. The criteria for a diagnosis of cEDS are broken down into two categories: major diagnostic criteria and minor diagnostic criteria. Major criteria include overly stretchy skin, atrophic scarring, and generalized joint hypermobility (this depends on age, gender, and family/ethnic background). Minor criteria include:
- Easy bruising
- Soft, doughy skin
- Fragile skin (breaks apart easily)
- Fleshy lesions called molluscoid pseudotumors that are associated with scars of pressure points such as your elbows and knees
- Small round hard masses called subcutaneous spheroids that are usually under the skin around the forearms and shins
- History of a hernia
- Epicanthal folds
- Complications associated with joint hypermobility (chronic pain, dislocations, sprains, etc)
- Family history of a first-degree relative (parent, sibling, child) who meets criteria for cEDS
Red flags that can increase the chance for someone to have cEDS would be if they have the major criteria of overly stretchy skin and atrophic scarring, or if they have the major criteria of generalized joint hypermobility combined with three or more of the minor criteria. Anyone who feels that they meet these criteria should seek consultation with a geneticist, genetic counselor, or other provider who is familiar with cEDS to get more information.
Medical Management for Classical Ehlers-Danlos syndrome
Treatment for cEDS can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with cEDS. Medical management can include blood tests, medications, physical therapy, and treatments to help with excessive bleeding or skin breakage. Imaging of the heart, called an echocardiogram may also be done to look for dilation of the aorta or mitral valve prolapse. It is also recommended that individuals with cEDS avoid unnecessary trauma and make sure any wounds are being properly cared for. Young children with skin fragility may want to wear pads or bandages to avoid skin tears, and all individuals with cEDS should avoid sports with heavy strain on the joints (such as contact sports, running, etc).
For women, during pregnancy they should be followed by a high-risk OB/GYN due to potential complications both for mom and the baby.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Loeys Dietz syndrome is a genetic condition that affects connective tissue. Connective tissue is a type of tissue that helps to hold everything together, like a glue for your body. The main body systems affected by Loeys Dietz syndrome include cardiovascular, skeletal, cutaneous (skin), and ocular (eyes):
- Cardiovascular – The most common cardiac findings associated with Loeys Dietz syndrome are congenital (from birth) heart defects, such as patent ductus arteriosus (PDA), atrial or ventricular septal defects (ASD/VSD), and bicuspid aortic valve (BAV).
- Skeletal – Patients with Loeys Dietz syndrome can have long fingers and toes, scoliosis, joint laxity, chest wall deformities (pectus excavatum or pectus carinatum, and/or osteoarthritis, as well as craniofacial findings such as flat cheek bones, downward-slanting eyes, craniosynostosis, cleft palate, and chin deformities (such as a small, receding chin).
- Cutaneous – Skin findings associated with Loeys Dietz syndrome include soft, velvety, translucent skin, easy bruising, abnormal scarring, and hernias.
- Ocular – Eye issues seen in patients with Loeys Dietz syndrome include myopia (nearsightedness), eye muscle disorders, and retinal detachment.
Additional unique clinical findings include arterial tortuosity (twisting arteries), hypertelorism (widely spaced eyes), bifid or broad uvula, and aortic aneurysms and dissections. Both aortic aneurysms and dissections can be life threatening. Some patients with Loeys Dietz syndrome also have food allergies, asthma, eczema, esophagitis/gastritis, and/or inflammatory bowel disease.
There are five types of Loeys Dietz syndrome which are associated with changes in different genes: I (TGFBR1), II (TGFBR2), III (SMAD3), IV (TGFB2), and V (TGFB3). Loeys Dietz syndrome can range greatly in severity, and there can be much variability even between affected members of the same family. Individuals may have few or no symptoms while others may have severe and early-onset health concerns.
Causes
Loeys Dietz syndrome is caused by pathogenic variants in the TGFBR1, TGFBR2, SMAD3, TGFB2, or TGFB3 genes, which are inherited in an autosomal dominant pattern. This means that a single copy of the pathogenic variant is enough to cause an individual to develop the condition, and anyone who carries the pathogenic variant has a 50% chance to pass it down to any children they have. About 25% of people who have Loeys Dietz syndrome have a parent who also has it. The other 75% are the first ones in their family to have it (called de novo).
Diagnosing Loeys Dietz syndrome
While genetic testing for Loeys Dietz syndrome can be helpful, medical providers may use other pieces of information, such as a physical exam and family history, to help establish the diagnosis. Some red flags that can increase the chance for Loeys Dietz syndrome in a family include:
- Aortic root enlargement or dissection
- Arterial tortuosity in the head and neck vessels
- Aneurysms
- Characteristic craniofacial, skeletal, cutaneous, and vascular features
- Family history of health concerns related to Loeys Dietz syndrome, or a family history of a pathogenic variant in the TGFBR2, SMAD3, TGFB2, or TGFB3 genes.
Due to the varying types of genetic changes that have been reported in the TGFBR1, TGFBR2, SMAD3, TGFB2, or TGFB3 genes in association with Loeys Dietz syndrome, genetic testing for Loeys Dietz syndrome involves sequence and deletion/duplication analysis. About 90-95% of those who meet the criteria for Loeys Dietz syndrome based on their personal and family history will have a pathogenic variant in one of the known causative genes. This also means that some individuals who appear to have Loeys Dietz syndrome will have negative genetic test results.
Medical Management for Loeys Dietz syndrome
Treatment for Loeys Dietz syndrome can sometimes vary depending on the individual person and their specific health concerns and should be discussed with a medical provider who is familiar with Loeys Dietz syndrome. Management for Loeys Dietz syndrome is generally focused on monitoring for the potential health complications. Individuals with Loeys Dietz syndrome are recommended to have regular eye exams by an ophthalmologist, annual cardiac imaging to monitor the aorta (screening may be more frequent if dilation is found), and intervention and follow-up by an orthopedist for those with progressive scoliosis or other skeletal problems. Additional screening may be recommended depending on the individual’s personal and family history. Because of the cardiovascular and other risks, it is often suggested that individuals with Loey Dietz syndrome avoid certain activities such as contact and competitive sports. It is also important for women with Loeys Dietz syndrome who are considering pregnancy to discuss options and review these risks beforehand when possible.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Marfan syndrome is a genetic condition that affects connective tissue. Connective tissue is a type of tissue that helps to hold everything together, like a glue for your body. The main body systems affected by Marfan syndrome include the eyes (ocular), bone and joints (skeletal), and heart and blood vessels (cardiovascular).
- Ocular: The most common eye findings are severe nearsightedness (also called myopia) and displacement of the lens of the eye (also called ectopia lentis). Individuals with Marfan syndrome may also be at risk for early cataracts, early glaucoma, and retinal detachment.
- Skeletal: Common findings include long arms and legs, long fingers and toes (also called arachnodactyly), a tall and thin body type, curvature of the spine (scoliosis), chest wall abnormalities (called pectus excavatum or carinatum), flat feet, high palate, and joint laxity (overly flexible).
- Cardiovascular: Differences with the heart and major blood vessels of the body are leading causes of serious health problems for people with Marfan syndrome. One of the biggest complications is called dilation of the aorta, which is the stretching of the main blood vessel that carries blood from the heart to the body. Dilation of the aorta can lead to an aneurysm (bulge in the aorta) or dissection (tear in the aorta). Both aortic aneurysms and dissections can be life threatening. Another heart problem, called mitral valve prolapse (MVP), is also more common in people with Marfan syndrome.
Additional things that can be seen in association with Marfan syndrome include collapsed lung (also called spontaneous pneumothorax), enlargement of the membrane around the brain and spinal cord (also called dural ectasia) which can cause pain, respiratory problems such as asthma or emphysema, stretch marks, and other health concerns. Some individuals may have learning difficulties, but this is rare.
Marfan syndrome can range in severity, and there can be variability within a family. Individuals may have few or no symptoms while others may have severe and early-onset health concerns.
Causes
Marfan syndrome is caused by pathogenic variants in the FBN1 gene, which are inherited in an autosomal dominant pattern. This means that a single copy of the pathogenic variant is enough cause an individual to develop the condition, and anyone who carries the pathogenic variant has a 50% chance to pass it down to any children they have. About 75% of people who have Marfan syndrome have a parent who also has it. The other 25% are the first ones in their family to have it (called de novo). Marfan syndrome affects 1 in every 5,000-10,000 people.
Diagnosing Marfan syndrome
While genetic testing for Marfan syndrome can be helpful, medical providers may use other pieces of information, such as a physical exam and family history, to help establish the diagnosis. Some red flags that can increase the chance for Marfan syndrome in a family include:
- Dilation of the aorta
- Ectopia lentis (dislocation of the lens of the eye)
- Systemic score (takes into account skeletal and other features)
- Family history of health concerns related to Marfan syndrome, or a family history of a pathogenic variant in the FBN1 gene.
These criteria are called the “Ghent nosology” and the Marfan Foundation has created an online tool that incorporates these criteria.
Due to the varying types of genetic changes that have been reported in the FBN1 gene in association with Marfan syndrome, genetic testing for Marfan syndrome involves sequence and deletion/duplication analysis. About 70-95% of those who meet the criteria for Marfan syndrome based on their personal and family history will have a pathogenic variant in the FBN1 gene.
Medical Management for Marfan syndrome
Treatment for Marfan syndrome can sometimes vary depending on the individual person and their specific health concerns, and should be discussed with a medical provider who is familiar with Marfan syndrome. Management for Marfan syndrome is generally focused on monitoring for the potential health complications. Individuals with Marfan syndrome are recommended to have regular eye exams by an ophthalmologist, annual cardiac imaging to monitor the aorta (screening may be more frequent if dilation is found), and intervention and follow-up by an orthopedist for those with progressive scoliosis or other skeletal problems. Additional screening may be recommended depending on the individual’s personal and family history. Because of the cardiovascular and other risks, it is often suggested that individuals with Marfan syndrome avoid certain activities such as contact and competitive sports. Because of the potential risks, it is important for women with Marfan syndrome who are considering pregnancy to discuss options and review these risks beforehand when possible.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Genetics & Personalized Medicine
Precision medicine is often used interchangeably with “personalized medicine” and “individualized medicine.” All of these terms describe an approach to healthcare that focuses on individual differences in genetics, environment, and lifestyle rather than a “one size fits all” kind of care. One aspect of precision medicine uses tests for certain biomarkers, like genetic variations, that will help to identify which groups of people will benefit from which targeted treatments and prevention plans.
Only recently has technology advanced enough to allow researchers to have the ability to test for these genetic variations. Some of these tests involve one or a few genes (genetic testing) or many or all of the genes (whole genome testing). While whole-genome testing is available, the interpretation remains very complex and carries with it the potential for a lot of uncertainty.
However, the National Institutes of Health (NIH) has been charged with a long-term research endeavor called the Precision Medicine Initiative to help us learn how this kind of care can help society. With the information generated from this research, we can begin to understand how individual differences in lifestyle, environment, and genetics can change disease expression.
Most precision medicine genetic testing involves panels of genes. This means there are several genes that may fall into different categories, but it’s important to realize that not all of these genes have been thoroughly studied yet and may not provide actionable information. Some laboratories will offer the testing by category, some will offer combinations of categories, and some will offer it as a comprehensive test covering many different health interests. Much of the research in the field is very new and it may be wise to have some caution. We encourage you to discuss it more thoroughly with your care team or an expert.
As with other types of genetic testing, it is important for you to consider the implications of precision medicine information. Deciding on whether or not to have any genetic test is a very personal decision. Some people choose to perform genetic testing because they feel it will empower them to make informed decisions that will lower the risk for disease. Some people do not feel that they are ready to know what could be revealed by genetic testing. This can be due to many different reasons, including feeling overwhelmed by other health concerns, feeling that the test results would not affect how they approach their medical care, or not wanting to know about risks that have no treatment options. These are all important issues to consider when deciding about precision medicine genetic testing.
Genetic Support Foundation is a resource for you – translating the science and helping you understand what it means for you and your family. We have information on a variety of different genetic tests that are currently on the market, but as this is a rapidly changing landscape, we will continue to keep you updated on many new and exciting discoveries generated by important studies like the Precision Medicine Initiative.
We have broken down the different categories of precision medicine genetic testing; please keep in mind that some of these tests are for informational purposes and not recommended for altering medical decisions.
We often like to say cancer is always genetic but not always hereditary. What we mean is that cancer develops as a result of accumulated DNA damage, also called variants. Most of the time those variants are acquired from a variety of different sources over the course of a lifetime like environmental exposures, lifestyle habits, occupational hazards, or certain types of viruses. Sometimes these variants happen by random chance. Acquired variants are also called somatic variants, or somatic mutations.
Cancer occurs because these DNA variants cause a cell to quickly grow and divide without normal cellular regulation while continuing to accumulate more and more genetic variants. At some point, this leads to formation of cancer (or a tumor). Because tumor cells have acquired so many genetic variants along the way, the sequence of DNA isolated from a person’s tumor can be very different from that individual’s inherited DNA.
In some cases, testing for the specific variants that are driving a cancer can help to determine what treatments would be best to use. More and more therapies are being developed that can more accurately target these variants that are specific to one cancer, which often results in better outcomes with fewer side effects.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Predictive tests can provide information about how a patient may respond (or be resistant) to treatment. Some DNA variants that lead to cancer also make the cancer cells susceptible to the effects of certain drugs. These drugs are called targeted therapies, because they target the genetic changes as a way of fighting the cancer. The targeted therapy is specifically designed for a particular pathway in the cancer cell making it more likely than other non-targeted treatment options to kill the cancer cells. Genomic testing of the tumor tissue (also called tumor profiling), which is a predictive test, can involve one specific tumor gene or several tumor genes depending on validated and repeated trials. Targeted therapies are available for colorectal, lung, and breast cancers, and a few others.
Unfortunately, even the most promising targeted therapies that report a highly statistically significant and clinically relevant reduction in the risk of disease events are unlikely to benefit all or even most patients. Large scale studies are needed to understand the incredible variability of cancer disease expression. The use of predictive genomic testing clearly has enormous clinical potential to help very rare, select individuals, but more information is needed to apply this in a larger population.
The term predictive testing may also refer to predisposition testing which is different because it looks at the inherited (or germline) DNA.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
In cancer, most prognostic tests are biomarkers that help indicate likelihood of disease events such as progression of disease, metastasis (spread of cancer), or recurrence. Prognostic markers can also be used to predict which patients will have a very low risk of disease events and can avoid toxic treatment, or high-risk patients who may benefit from more aggressive therapies. While presence or absence of a prognostic marker can be useful to determine which patients need treatment it does not directly predict the response to a treatment.
Biomarkers can be lymph nodes, pathology features, or proteins found in the blood. Recently genetic variants in tumor cells have also been included with the prognostic tools used in cancer treatment. These tests examine several genes to help determine the likelihood of recurrence and whether that patient should continue chemotherapy.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Direct-to-consumer (DTC) testing is a fairly new category of testing in which genetic tests are made available to the general public without requiring an ordering healthcare provider or insurance coverage. Saliva specimen collection kits are mailed to a consumer who sends it back to a laboratory, and then receives a report on a variety of different interests including ancestry information, non-health related traits, and other topics. DTC technology often does not use the same validation process for DNA changes as other medical genetic testing, and thus it is difficult to know for sure how accurate they are. Several genetics professional societies have issued statements about these kinds of tests here so it is important for you to make an informed decision about what kind of genetic test is most beneficial for you.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
While most screening tests we talk about during pregnancy are designed to give us more information about the baby, genetic carrier screening is a test that gives us information about mom and dad.
As you may recall from Genetics 101, we have two copies of most genes: one we get from our mom and one we get from our dad. When we say that someone is a ‘carrier’ for a genetic condition, it means that there is a harmful change (called a pathogenic variant) in one of the copies of the gene that make it not work how it should. Because there is still one gene that is working properly, carriers generally do not show signs or symptoms of any health issues related to being a carrier.
Genetic carrier screening usually looks for conditions that are recessive, meaning that for someone to have the genetic condition both copies of the gene need to be not working. In most instances, if someone is a carrier for a recessive condition, they don’t have any medical or health problems, and may not have a family history of any genetic conditions.
Because being a carrier for a genetic condition does not cause any health problems in most cases, then what’s the point of testing for it? We are all probably carrier for five or more recessive genetic conditions. If two people who are carriers for the same genetic condition have a child, there is a 25% chance that they will have a child that has that genetic condition. This means there is a 1 in 4 chance that each parent will pass down their non-working copy of the gene to the pregnancy, and then the baby will have no working copies of that gene.
Carrier screening may also offer testing for X-linked recessive conditions. X-linked means that the gene is located on the X chromosome. To review from Genetics 101, women have two X chromosomes and men have one X and one Y chromosome (and thus only one copy of all of the genes on the X chromosome). If a male inherits a nonworking gene on his X chromosome, he usually will have that genetic condition because he does not have a second working copy of that gene.
If a woman, on the other hand, inherits a nonworking gene on one of her X chromosomes, she usually has a working copy of that gene on her other X chromosome. If a woman is a carrier for an X-linked recessive condition, she may have very mild or no symptoms at all. A woman who is a carrier for an X-linked recessive genetic condition would have a chance to pass that condition down to her children. Click here to see a list of commonly tested conditions.
How it Works
Genetic carrier screening can generally be done by either a blood or a saliva test. The blood or saliva is sent to the lab, where they can then pull out your DNA from either your blood cells or your cheek cells that are in your saliva. This DNA is what makes up your genes. Your genes are basically a string of letters that are an instruction for how to make a protein. Each protein has a very specific job to do in the body. Some proteins tell our body how tall we’re going to be, while others tell our eyes what color they will be.
Some proteins perform such important jobs in the body, that when we’re born without them, they can cause health problems, known as genetic conditions. If there is a harmful spelling error in the DNA, known as a pathogenic variant, it can change the instructions for that gene. If the instructions are changed, then that gene can make either an abnormal protein or no protein at all. Not having these proteins can lead to different health concerns. To learn about the different kinds of pathogenic variants, see sequencing and deletion/duplication testing.
Each lab offers screening for various genetic conditions. Most of the conditions that are offered for prenatal, or preconception carrier screening are passed down in a family in a pattern we call autosomal recessive, although there are some X-linked conditions that can be screened for.
Expanded Carrier Screening
Some medical providers offer expanded genetic carrier screening. Expanded carrier screening offers testing for many genetic conditions at one time. There are many viewpoints and ethical issues involved with expanded carrier screening, and like most things there are pros and cons.
Expanded carrier screening is available to all individuals, regardless of ethnicity, but your insurance may or may not cover this testing. Some of the conditions that expanded carrier screening looks for are severe, while others may be milder, or not even have any significant medical concerns associated with it. Some of these conditions have available treatments, while others don’t.
There is also a very small chance that you could find out that you actually have a genetic condition, depending on the genetic conditions that are screened for.
Labs that offer expanded carrier screening may or may not be as thorough as labs that do not focus on expanded carrier screening. For example, if you have a family history of a specific condition, expanded carrier screening may not be the best test for you.
Whether or not to move forward with expanded carrier screening can be a complicated decision, and it is important to discuss it with a medical provider, such as a genetic counselor, to fully understand what the test is able to tell you.
How to Decide
The choice of whether or not to undergo genetic carrier screening is yours. There may be some questions that would be helpful to consider before you make the decision on whether or not to pursue carrier screening:
- Would I like to know as much information as possible to plan and be prepared?
- Would having more information make me more anxious or nervous?
- If testing on my partner and I indicated an increased risk for a genetic condition, would I want to move forward with diagnostic testing (such as CVS or amniocentesis), that would be able to give a ‘yes’ or ‘no’ answer, but also carries a risk for miscarriage?
- If I knew that my baby had one of these conditions, would it affect my decision to continue the pregnancy? Would I consider alternative options, such as terminating the pregnancy, or placing the baby up for adoption?
This is not an inclusive list of things to consider but may be a good place to start. It is important to also discuss what sort of screening would be right for you with your doctor or genetic counselor.
Carrier Screening Results
My results were normal/negative. What does that mean?
When it comes to genetic carrier screening, a negative result significantly reduces, but does not eliminate, the chance to be a carrier of those genetic conditions. The chance to be a carrier for these genetic conditions after a negative genetic test is called the residual risk. The residual risk will vary depending on the laboratory that runs the test and the type of testing that was performed. Your provider or genetic counselor can help you determine what your residual risk is after having a negative genetic carrier screening test.
My results were positive, or said that I’m a carrier for a genetic condition. Now what?
To be a carrier for a genetic condition generally means that you have one working and one non-working copy of a gene. Most of these genes follow autosomal recessive inheritance patterns, which mean that someone would have to inherit two non-working copies of a gene in order to develop that genetic condition. For example, if someone inherits a non-working copy of the CFTR gene from both parents, it causes cystic fibrosis. However, if someone carries one non-working copy of a gene, they generally do not have any symptoms. However, they do have a higher chance to have a child that may have that genetic condition.
It is not uncommon to be a carrier for a genetic condition. We have over 20,000 genes, so everyone is likely a carrier for something. The next steps somewhat depend on what condition you were found to be a carrier for. If you were a carrier for a condition with autosomal recessive inheritance, the next step would be to consider carrier testing for your partner to see if they are also a carrier for the same genetic condition. If you and your partner are both found to be carriers for the same autosomal recessive genetic condition, there is a 25% chance for both parents to pass down the non-working copy of the gene, meaning that the baby would have that genetic condition.
If you are found to be a carrier for an X-linked condition, for women the chance to have an affected baby depends on whether the baby is a boy or a girl. If a woman is a carrier for an X-linked condition and is having a boy, there is a 50% chance to pass down the X chromosome with the nonworking gene, which would result in the baby having the genetic condition. There is also a 50% chance to pass down the X chromosome that has the working copy of the gene, which would result in a baby boy that does not have the genetic condition.
If the woman is a carrier for an X-linked condition and is having a girl, there is a 50% chance to pass down the X chromosome with the nonworking gene, which would mean that the baby girl would be a carrier for that genetic condition just like her mom. There is also a 50% chance to pass down the X chromosome that has the working copy of the gene, which would result in a baby girl that is not affected with the genetic condition and is also not a carrier for the genetic condition.
Both my partner and I are carriers of the same autosomal recessive genetic condition; what does this mean for our baby/potential baby and what are our options?
If you and your partner are both found to be carriers for the same genetic condition, there is a 25% (or 1 in 4) chance that the baby will inherit a non-working copy of the gene from both parents. That also means that there is a 50% chance that the baby will inherit one working and one non-working copy of the gene (which would make them carriers, just like their parents). There would also be a 25% chance that the baby will inherit a working copy of the gene from both parents.
Finding out that you and your partner are both carriers for the same autosomal recessive condition can be stressful. There are a couple of different options moving forward: either have prenatal diagnostic testing to find out of the baby is affected or wait until the baby is born and then revisit the option of genetic testing.
Prenatal diagnostic testing, such as chorionic villus sampling (CVS) and aminocentesis, comes with some risk (up to a 1 in 200 (0.5%) to 1 in 300 (0.03%) chance for a complication that could lead to a miscarriage from the procedure), but can provide a ‘yes’ or ‘no’ answer as to whether the baby is affected or not. For most genetic conditions, diagnostic testing can be done after the baby is born. The decision of whether to move forward with prenatal diagnostic testing is very personal and should be done after a talking through the risks, benefits, and limitations with your provider.
I found out that my partner and I are both carriers for the same recessive genetic condition and are concerned about having a child with a genetic condition. What are my options?
The answer to this question may depend on many things, including how severe the condition is, and what your personal thoughts are around having a child with a genetic condition. If you find that you and your partner are carriers for the same recessive genetic condition, there are several options:
- Move forward with pregnancy (or future pregnancy) as planned, and decline further genetic testing
- Move forward with pregnancy (or future pregnancy) and undergo diagnostic testing, such as CVS or amniocentesis, to find out during the pregnancy if the baby has the genetic condition. Some people want to pursue prenatal genetic diagnosis to be prepared for when the baby arrives, while other people want prenatal diagnosis because they would end a pregnancy that was affected. Other people would make plans for adoption if they found out the baby had a genetic condition. No matter the reason, it is important to discuss the risks, benefits, and limitations of diagnostic testing with your provider.
- Pursue in-vitro fertilization (IVF) with preimplantation genetic diagnosis (PGD). IVF with PGD involves fertilizing an embryo in a the lab, and then testing it to see if it has that genetic condition. You would then have the option to only transfer embryos that were not affected with the genetic condition. This process can be expensive, but some insurances may cover part or all of it.
- Consider using a sperm/egg donor
- Consider adoption
These options are not all easy, some of them cost a lot of money, and some of them may not fit with your personal beliefs and values. In terms of emotional support, it may be helpful to connect with another couple who has gone through this process; your provider or genetic counselor may be able to help facilitate this.
Click here to learn more about scheduling a genetic counseling appointment for pregnancy-related questions here or click here to learn more about scheduling a genetic counseling appointment for infertility or preconception questions.
Our genetic make-up helps to determine how our body will respond to and process (metabolize) medications and drugs. By evaluating an individual’s genetic variation in specific genes we can sometimes determine if a medication will be effective or cause serious toxic side effects. This is a relatively new branch of precision medicine called pharmacogenomics or pharmacogenetics.
Several organizations (CPIC and PharmGKB) have been working to develop guidelines for when to do genetic testing and how to modify medication dosage when an individual carries a variant gene. However, there are many drug/gene pairs that do not yet have recommended guidelines so selecting a reliable laboratory is critical to getting useful information.
Also keep in mind that there are many factors that affect our response to medications. Some of these are intrinsic, meaning that they are factors that are inherently part of us or controlled by our body. Examples of these are age, gender, race, and genetics. Some factors that affect our response to drugs are extrinsic, meaning that they come from outside of our body. Examples of these are diet, nicotine or alcohol use, and other drugs that we take. So it is important to remember that a genetic test may still not give you the whole story.
There are, however, some underlying genetic conditions that can have a very clear and direct impact on how someone will react to a type of medication, such as malignant hyperthermia susceptibility (MHS).
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
There are three different types of technology used for ancestry testing: Y-DNA, mitochondrial (mt) DNA, and autosomal DNA. Males carry an X and a Y chromosome, whereas females carry two X chromosomes. Males will give their sons the Y chromosome and their daughters the X chromosome which means a Y chromosome DNA test will trace ancestry through the male side of the family. Mitochondrial DNA is present in both males and females, but only mother’s pass on the mtDNA. This means mtDNA testing will trace ancestry through the female side of the family. Both of these tests try to identify a single line of descent (called a haplotype) dating back thousands of years. Autosomal DNA comes from both male and female sides of the family. In this case the lab analyzes a number of specific locations throughout the individual genome.
Ancestry genetic testing is not quite the same as medical genetic testing. The lab will compare the consumer’s DNA to a reference database of DNA samples from modern individuals living in various regions. Keep in mind that there are disagreements between the definitions of certain genetic ethnicities, many ethnic groups migrated from their country of origin, and the accuracy of the test is directly related to the variety of ancestries represented in the databases. In other words the labs use statistics and prediction models to give you a probability of ethnicity, but there are many explanations for why it may not match up with what you expect.
Click here to learn more about scheduling a genetic counseling appointment for questions about pediatric or adult genetic conditions.
Have questions?
Do you have questions about pediatric or adult genetic conditions? Talk with one of our genetic counselors.
NINDS conducts, fosters, and supports research and research training on the causes, prevention, diagnosis, and treatment of neurological and muscle disorders.
Genetic brain disorders affect the development and function of the brain. Some are inherited, some are caused by exposure, and others are both. Learn types and treatments.
Brain malformations is damage or abnormal development of the brain and nervous system. Starts long before a baby is born. Learn types and treatments.